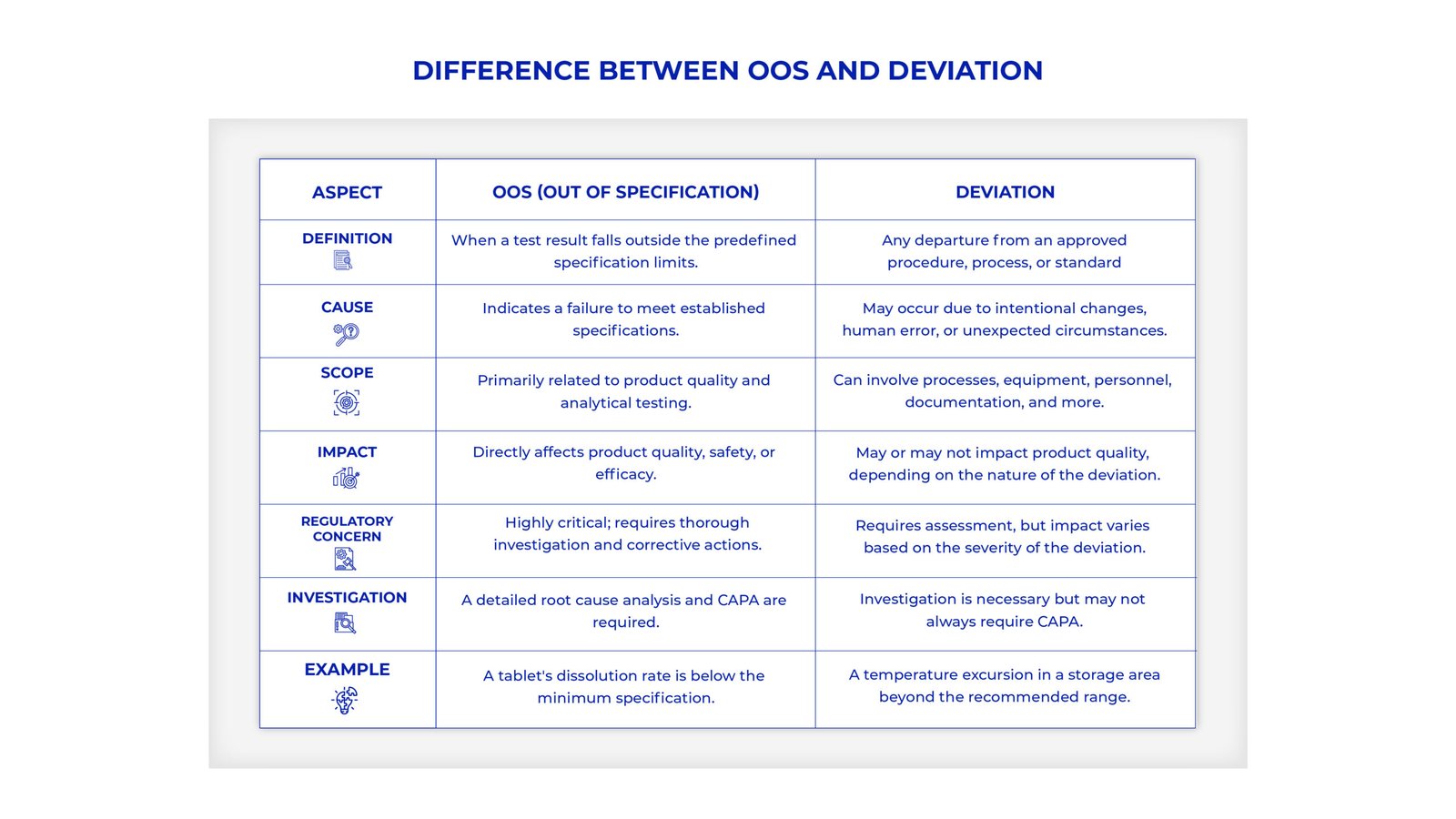
A systematic and disciplined methodology for OOS investigations is crucial to discovering root causes and developing solutions. Here is a step-by-step investigation procedure for a solid OOS investigation framework:
Phase 1: Initial Laboratory Investigation
- Initial Laboratory Assessment
- Data Verification: Confirm the accuracy of the test data, ensuring proper instrument calibration and adherence to analytical methods.
- Analyst Review: Evaluate analyst performance to rule out human error, including potential sample mishandling or miscalculations.
- Expanded Laboratory Investigation
- Re-testing Protocol: Conduct re-testing to determine if the OOS result persists, ensuring that re-testing is justified and scientifically valid.
- Methodology Review: Assess the suitability and robustness of the analytical methods employed, making adjustments as necessary.
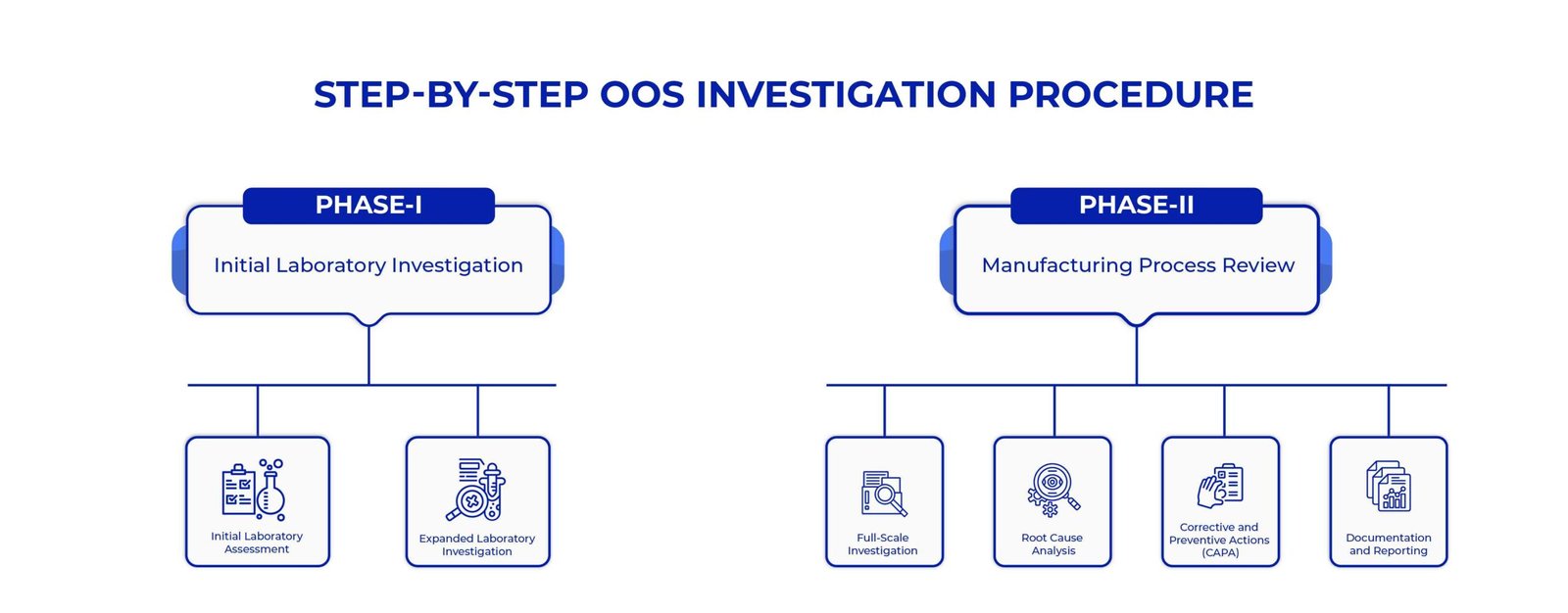
Phase 2: Manufacturing Process Review
- Full-Scale Investigation
- Production Process Examination: Investigate manufacturing processes for deviations or anomalies that could contribute to OOS results.
- Material Quality Assessment: Evaluate raw materials and components for conformity to quality standards, identifying potential sources of variation.
- Root Cause Analysis
- Analytical Tools Application: Utilize tools such as Fishbone Diagrams, 5-Whys Analysis, or Failure Mode and Effects Analysis (FMEA) to pinpoint the underlying cause of the OOS result.
- Corrective and Preventive Actions (CAPA)
- Immediate Corrections: Address the identified root cause promptly to prevent recurrence.
- Preventive Strategies: Implement long-term measures, such as process optimisation or enhanced training programs, to mitigate future risks.
- Documentation and Reporting
- Comprehensive Record-Keeping: Document all aspects of the investigation, including findings, actions taken, and rationale for decisions, ensuring transparency and traceability.
- Regulatory Communication: Report significant findings to regulatory authorities as required, maintaining compliance with current guidelines.
AmpleLogic integrates with CAPA (Corrective and Preventive Action) and LIMS (Laboratory Information Management Systems) to streamline root cause analysis and ensure seamless cross-departmental collaboration during Phase II.